Introducción
Las lesiones de cabeza y cuello son un motivo de consulta frecuente en pediatría. La mayor parte de ellas corresponde a aumentos de volumen y generalmente con características de benignidad, relacionadas con infecciones, inflamaciones, acumulación de líquidos, o tumefacciones. Pese a esto, es importante considerar siempre una etiología maligna que detectada y manejada precozmente presenta un buen pronóstico.
El cáncer es la segunda causa de muerte infantil, destacando en frecuencia la leucemia, los tumores del sistema nervioso central y los linfomas, seguidos por los tumores sólidos como Tumor de Willms, los de tejidos blandos, los óseos, entre otros. Se ha estimado que 5-10% de los tumores malignos primarios en niños se producen en cabeza y cuello. Dentro de estos tumores destaca el rabdomiosarcoma infantil que representa aproximadamente el 3,5% de los casos de cáncer en niños de 0 a 14 años de edad y 2% entre adolescentes y adultos jóvenes entre los 15 y los 19 años de edad.
Este artículo es la exposición de un caso clínico de rabdomiosarcoma infantil cuya forma de presentación puede confundir al médico general, retardar el diagnóstico y como consecuencia de ello, comprometer el pronóstico de la enfermedad.
Definición
El rabdomiosarcoma es un tumor maligno de tejido blando de origen musculoesquelético que puede formarse virtualmente en cualquier parte del cuerpo (1). Es el tumor de partes blandas más frecuente en los niños, con una incidencia de un 38% en cabeza y cuello. El rabdomiosarcoma más frecuente es el embrionario y se presenta generalmente en niños menores de 6 seis años de edad. Por otro lado, el alveolar se observa en niños mayores de 6 años, y representa alrededor del 20 por ciento de la totalidad de los casos. Se ha demostrado que en cabeza y cuello el rabdomiosarcoma embrionario es discretamente más frecuente que el alveolar (2).
La mayoría de los casos se presenta de forma esporádica sin ningún factor de riesgo o de predisposición reconocible, a pesar que una pequeña porción de estos están relacionados con factores genéticos. Es una enfermedad curable en la mayoría de los niños que reciben terapia de modalidad combinada, con una supervivencia de más del 70% a los 5 años después de realizado el diagnóstico. (3)
Presentación Clínica
Los síntomas y signos son variados, dependiendo del tamaño y la localización del tumor. Así, los tumores en la nariz o la garganta pueden producir sangrado, congestión, dificultades en la deglución o problemas neurológicos cuando se extienden hasta el cerebro. Cuando estos tumores se presentan en los músculos pueden aparecer como protuberancias dolorosas y a menudo se cree que son lesiones de tipo benignas. Los síntomas también pueden no manifestarse hasta que el tumor está muy desarrollado, produciendo un retraso en el diagnóstico y en el tratamiento.
Entre los síntomas y signos más comunes destacan:
- Tumor o masa visible o palpable, con o sin dolor
- Sangrado de la nariz o la garganta.
- Parestesias, si el tumor comprime los nervios del área donde se sitúa.
- Una protuberancia en el ojo o ptosis palpebral, lo que indicaría la presencia de un tumor en el globo ocular.
Diagnóstico
Los síntomas del rabdomiosarcoma pueden parecerse a los de otras condiciones o problemas médicos, por lo que el examen físico y la historia médica completa son de vital importancia para llegar a un diagnóstico certero y oportuno. Además, existen métodos de laboratorio que nos sirven para confirmarlo.
Para comenzar el estudio debemos realizar:
·Biopsia del tumor.
·Análisis de sangre y de orina.
·Imagenología: La elección del método a utilizar dependerá de las condiciones clínicas del paciente, la ubicación topográfica del aumento de volumen, y la información que queramos obtener. Entre ellas destacan: tomografía axial computarizada, resonancia nuclear magnética (RNM), radiografía, ecografía y cintigrama óseo. Es interesante destacar el reciente uso de la RNM en el diagnóstico prenatal del rabdomiosarcoma fetal (4).
Una vez confirmado el diagnóstico, se determina la etapa en la que se encuentra la enfermedad, lo que es fundamental para definir el pronóstico y tratamiento a seguir.
Uno de los métodos usados para etapificar es el TNM, que se basa en el tamaño y ubicación del tumor (T), el estado de los nódulos (N) y la presencia o ausencia de metástasis (M). A través de este sistema se realiza una evaluación de la enfermedad previa a la cirugía y se la clasifica en una de las cuatro etapas siguientes:
Etapa I: El tumor se encuentra en el globo ocular, la cabeza, el cuello o el tracto genitourinario (excepto la próstata y la vejiga). El tumor está localizado, lo que significa que no se ha diseminado a otras áreas del cuerpo.
Etapa II: El tumor está localizado en un área solamente (ninguna de las áreas de la etapa 1), es pequeño y mide menos de 5 cm. Las células del tumor no se han diseminado a los ganglios linfáticos circundantes.
Etapa III: El tumor está localizado en cualquier área no incluida en la etapa 1, mide más de 5 cm. y puede o no haberse diseminado a los ganglios linfáticos circundantes.
Etapa IV: El tumor se encuentra diseminado a otras áreas del cuerpo en el momento del diagnóstico.
Tratamiento
El tratamiento del rabdomiosarcoma se basa en el control local y a distancia del tumor. Para esto se necesita un manejo multidisciplinario que incluye cirugía, quimioterapia y radioterapia (5). El tipo de tratamiento depende de la histología del
tumor, de la edad del paciente, la localización, el tamaño y la etapa preoperatoria (6). En Chile el PINDA (Programa Infantil Nacional de Drogas Antineoplásicas), siguiendo los protocolos del IRSG (Intergroup Rhabdomyosarcoma Study Group), está encargado de coordinar el tratamiento de los pacientes.
Actualmente existen 3 pilares en el tratamiento:
Cirugía: Extirpación del tumor con bordes libres, tanto como sea posible, sin que la cirugía sea mutiladora, obteniendo una muestra de los nódulos linfáticos de drenaje de la zona. Puede ser necesario realizar varias cirugías dependiendo de la evolución del paciente.
Quimioterapia: su objetivo es prevenir la diseminación posterior del tumor. Entre los medicamentos se encuentran: Vincristina, Actinomicina–D, Ciclofosfamida, Doxorubicina, Melfalán, Ifosfamida, Etopósido y Topotecan. Además puede ser usado como tratamiento complementario a la cirugía, administrándose por 10 a 12 meses, previo a ésta, para reducir el tumor. (7).
Radioterapia: está indicada en aproximadamente el 90% de los casos y juega un rol importante en el control local de la enfermedad y en aumentar la sobrevida. Actualmente se ha desarrollado la radioterapia conformacional externa, que tiene como objetivo aumentar las dosis de irradiación al tejido tumoral, disminuyéndola a los tejidos sanos, para evitar efectos secundarios no deseados como alteraciones en el crecimiento y desarrollo y el riesgo potencial de formación de neoplasia secundaria, que tiene la radioterapia convencional. Los pacientes libres de evento a 5 años varían de 36% cuando se usa una técnica de radioterapia convencional a 71% cuando se usa una técnica conformacional (8). En términos de sobrevida global a 5 años, ésta es de 55% y 74%, respectivamente.
Pronóstico
El pronóstico para un niño o adolescente con rabdomiosarcoma se relaciona con lo avanzado de la enfermedad, tamaño y ubicación del tumor, presencia y número de metástasis, respuesta del tumor a la terapia, edad y estado general de salud del paciente y el grado e histología del tumor (9). Estudios han demostrado que la sobrevida a cinco años es mayor en niños entre 1-4 años, cuando la enfermedad es localizada y tiene histología embrionaria. Por el contrario un peor pronóstico se asocia a tumores diagnosticados adolescencia, tipo histológico alveolar, el mayor tiempo de enfermedad y a la presencia de metástasis. (10)
La atención médica precoz y una terapia intensa contribuyen a un mejor pronóstico. Una vez realizado el diagnóstico, el seguimiento continuo es esencial. Un paciente que sobrevive a un rabdomiosarcoma puede presentar evidencias de los efectos secundarios de la radioterapia y la quimioterapia. Además se debe tener presente que es posible la recidiva.
Caso Clínico
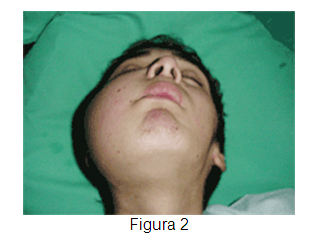
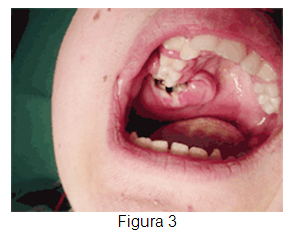
Paciente de 14 años, sexo masculino, consulta a odontólogo por cuadro de aproximadamente un mes de evolución, caracterizado por aumento de volumen leve en la mejilla derecha asociado a dolor (Figuras 1 y 2). El especialista diagnostica cuadro de origen infeccioso e indica tratamiento antibiótico. El menor consulta nuevamente 2 semanas después por aumento progresivo de volumen, intensificación del dolor y sangrado. Además se agrega lesión de tipo excrecente en la zona (Figura 3) por lo que el odontólogo decide realizar una biopsia por punción, la cual revela la presencia de células neoplásicas malignas.
El paciente es derivado a un Servicio de Oncología donde se realiza biopsia quirúrgica que demuestra la presencia de un rabdomiosarcoma. La ubicación de la lesión, en cara, hace necesaria la reducción de la masa mediante quimioterapia, previo a la extirpación quirúrgica para disminuir la agresividad de la cirugía.
Discusión
El cáncer en niños es una patología poco frecuente, sin embargo, es un diagnóstico diferencial que siempre debe ser considerado ya que la presentación clínica puede ser similar a una patología benigna, y su detección precoz y manejo oportuno le otorga un pronóstico favorable.
Conclusión
Las causas de muerte pediátrica en nuestro país corresponden en primer lugar a los accidentes y en segundo al cáncer, por lo tanto es una patología que debe estar siempre presente en la sospecha diagnóstica. La leucemia ocupa el primer lugar, seguida por tumores sólidos entre los que el rabdomiosarcoma se presenta con una de las mayores frecuencias.
El rabdomiosarcoma representa un tercio de los tumores malignos de cabeza y cuello por lo que se debe tener presente si al examen físico nos encontramos frente a una masa tumoral fija, lobulada e irregular.
Referencias
- Breitfeld P., Meyer W. Rhabdomyosarcoma: new windows of opportunity. Oncologist. 2005 Aug; 10(7): 518-27. [Citado 1 de abril 2006] Disponible en Internet http://theoncologist.alphamedpress.org/cgi/content/full/10/7/518
- Hicks J., Flaitz C. Rhabdomyosarcoma of the head and neck in children. Oral Oncol. 2002 Jul; 38(5): 450-9.
- Stevens M. Treatment for childhood rhabdomyosarcoma: the cost of cure. Lancet Oncol. 2005 Feb; 6(2): 77-84.
- O'Callaghan M., House M., Ebay S., Bhadelia R. Rhabdomyoma of the head and neck demonstrated by prenatal magnetic resonance imaging. J Comput Assist Tomogr. 2005 Jan-Feb; 29(1): 130-2.
- Schouwenburg P., Kupperman D., Bakker F., Blank L., de Boer HB, Voute T. New combined treatment of surgery, radiotherapy, and reconstruction in head and neck rhabdomyosarcoma in children: the AMORE protocol. Head Neck. 1998 Jul; 20(4): 283-92.
- Morikawa Y. Childhood rhabdomyosarcoma. Nippon Geka Gakkai Zasshi. 2005 Jul; 106(7): 431-6.
- Stevens M., Rey A., Bouvet N., Ellershaw C., Flamant F., Habrand J., Marsden B., Martelli H., Sanchez de Toledo J., Spicer R., Spooner D., Terrier-Lacombe M., van Unnik A., Oberlin O. Treatment of nonmetastatic rhabdomyosarcoma in childhood and adolescence: third study of the International Society of Paediatric Oncology--SIOP Malignant Mesenchymal Tumor 89. J Clin Oncol. 2005 Apr 20; 23(12): 2586-7.
- Wolden L., Wexler L., Kraus D., Laquaglia M., Lis E., Meyers P. Intensity-modulated radiotherapy for head-and-neck rhabdomyosarcoma. Int J Radiat Oncol Biol Phys. 2005 Oct 1; 63(2): 647-8; author reply 648-9.
- Simon J., Paulino A., Smith R., Buatti J. Prognostic factors in head and neck rhabdomyosarcoma. Head Neck. 2003. May; 25(5): 416-7.
- Punyko J., Mertens A., Baker K., Ness K., Robison L., Gurney J. Long-term survival probabilities for childhood rhabdomyosarcoma. A population-based evaluation. Cancer. 2005 Apr 1; 103(7): 1475-83.
|