Introducción
El carcinoma hepatocelular fibrolamelar (CHC-FL) es una variante rara del carcinoma hepatocelular, que ocurre con mayor incidencia en adultos jóvenes, por lo que su presentación en la edad pediátrica es poco frecuente, representando menos del 1% de las neoplasias hepáticas en este grupo etario. Generalmente debutan con un cuadro clínico insidioso de dolor abdominal, sensación de masa y síntomas constitucionales, en pacientes sin antecedentes de hepatopatía crónica (1,2). A continuación, se presenta un caso clínico en una paciente de 11 años, sin antecedentes personales conocidos diagnosticado como hallazgo incidental durante el enfoque de trauma abdominal.
Caso clínico
Paciente femenina de 11 años, sin antecedentes patológicos previos, quien ingresa al servicio de urgencias con cuadro clínico de 3 días de evolución consistente en dolor abdominal localizado en hipocondrio de predominio derecho, el cual inicia posterior a caída desde su bicicleta, obteniendo trauma con el manubrio de esta. Al ingreso, la paciente niega cualquier otra sintomatología sistémica o local. Al examen físico, como hallazgo patológico se observa palidez cutánea generalizada con abdomen distendido, poco depresible, sin signos de irritación peritoneal, con dolor a la palpación en hipocondrio derecho y hepatomegalia de aproximadamente 3 cm que durante estancia hospitalaria presenta aumento progresivo llegando a medir aproximadamente 9 cm.
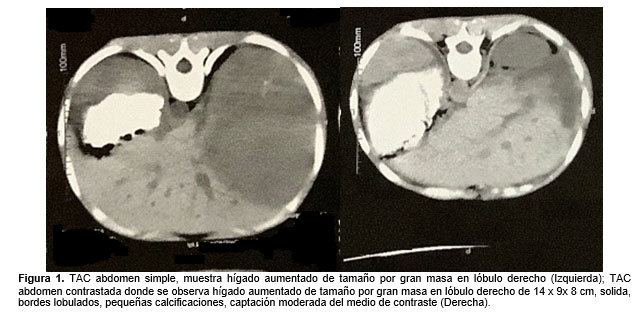
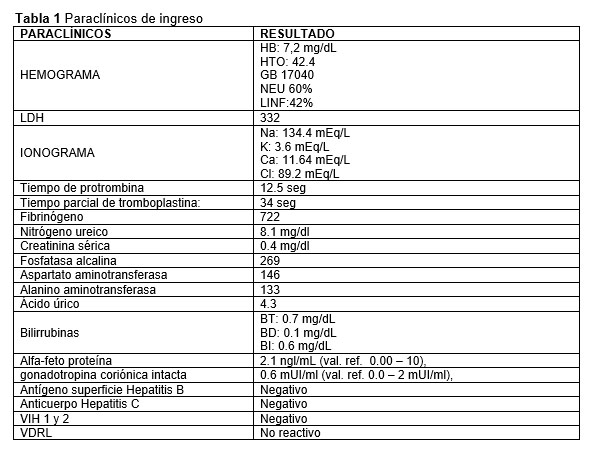
Los paraclínicos de ingreso se presentan en la tabla 1, llama la atención la presencia de leucocitosis, a expensas de neutrófilos, anemia leve sin requerimiento transfusional y trombocitosis; además de pruebas de función hepática ligeramente elevadas, sin datos de falla hepática. Se realizó ecografía de abdomen donde se evidenció lesión focal de aspecto multilobulado con flujo intraparenquimatoso y distorsión de contornos hepáticos, de aproximadamente 11 x 10 cm de diámetro, a partir de la cual, se estableció diagnóstico de neoplasia hepática y se inició estudios adicionales.
Por hallazgos, se realizó TAC de abdomen que reportó lesión tumoral hepática de gran tamaño (Figura 1), y RMN de abdomen simple y contrastada que reportó importante grado de hepatomegalia por presencia de gran masa tumoral que ocupaba la totalidad del lóbulo derecho con una longitud supero inferior de 16 cm, un diámetro transverso de 9.5 cm y una longitud antero posterior de 12.7 cm; con restricción de la difusión por alta celularidad y realce heterogéneo del material de contraste, especialmente en fase arterial y en menor grado en la fase venosa tardía (portal). Además, la lesión presentaba invasión e infiltración de las venas porta intrahepáticas, hallazgos altamente sugestivos de un hepatocarcinoma. Como complemento de estudios se realizó: TAC de tórax normal; ecocardiograma con dilatación leve de ventrículo izquierdo, insuficiencia plurivalvular leve y resto del estudio normal; y gammagrafía ósea normal.
Otros análisis serológicos realizados fueron marcadores tumorales y perfil infeccioso, todos dentro de rangos de normalidad (tabla 1). Se realiza junta médica entre los servicios de cirugía y oncología pediátrica y se decide realizar doppler para definir compromiso portal. En el reporte se evidenció una masa ligeramente hiperecogénica de contornos bien definidos ubicada en el segmento VI, de 85 mm de diámetro; y al examinar con Doppler se encuentra en sistema portal flujo fásico, en porta derecha e izquierda se encontraron permeables, sin embargo, la porta derecha se encontró disminuida en su diámetro en aproximadamente un 80% por efecto tumoral.
Con los hallazgos encontrados, se decidió que la paciente se beneficiaría de embolización arterial con esferas de PVA y COILS de masa hepática en segmentos V y VI, VII y VIII, la cual fue realizada por radiología intervencionista y posteriormente fue intervenida quirúrgicamente.
Durante el procedimiento quirúrgico, ingresan a cavidad peritoneal identificando hígado con gran masa tumoral en el lóbulo derecho y observaron una pequeña siembra tumoral en epiplón mayor, la cual fue resecada; posteriormente realizaron luxación hepática total y transección hepática a través del segmento lV, con lo cual realizaron una hepatectomía (trisegmentectomía) derecha ampliada ya que el tumor ocupaba los segmentos V,VI,VII y VIII, se encontraron lesión de 16 x 11 x 8.5 cm de color verde amarillento. La pieza resecada fue enviada a revisión por patología, quien emitió resultado con diagnóstico patológico de tumor epitelial primario, a favor de carcinoma hepatocelular fibrolamelar, bien diferenciado, unifocal y limitado al hígado. La paciente recibió alta hospitalaria con seguimiento ambulatorio por oncología y cirugía pediátrica.
Discusión
El carcinoma hepatocelular fibrolamelar es una variante infrecuente del carcinoma hepatocelular (CHC), representando el 0.85 a 16% de todos los CHC. Acontece en diferentes grupos etarios, pero usualmente no existe hepatopatía crónica de base (3), lo cual coincide con el caso reportado. Fue descrito en 1956 la primera vez por Edmondson, puede considerarse como una entidad diferente al CHC, ya que se asocia a pocos cambios cromosómicos y heterogeneidad genética, entre otras diferencias con este último (4). Afecta a ambos sexos por igual, prevalece en adultos jóvenes (<40 años), con una media de presentación a los 25 años, distinto a CHC donde la incidencia aumenta con la edad, con un pico a los 70 años (5).
El CHC-FL tiene como triada clásica en su presentación clínica la presencia de masa palpable en hipocondrio derecho, dolor abdominal y pérdida de peso. Se puede acompañar de síntomas inespecíficos como anorexia, náusea, fiebre o sensación de plenitud abdominal, y hasta el 40% de los pacientes debutan con ictericia, o manifestaciones poco frecuentes como síndrome de Budd Chiari, compromiso obstructivo biliar o falla hepática aguda (6) lo cual no se observó en el caso expuesto. A resaltar de este caso, el hallazgo incidental de esta variante rara de CHC fue secundario al proceso diagnóstico luego de un trauma abdominal en una paciente previamente asintomática. A diferencia de otros reportes en la literatura local, este caso se presentó en una edad menor y se identificó de forma temprana en el curso de su evolución, lo cual permitió indicar intervención quirúrgica. En otro reporte de caso de Latinoamérica, publicado por Téllez et al, se describió una paciente femenina de 17 años con CHC-FL típico, sin datos de hepatopatía crónica ni identificación de virus hepatotropos subyacentes, quien ingresó por síntomas crónicos de dolor abdominal localizado en epigastrio, masa en crecimiento progresivo y otros síntomas inespecíficos. Debido al cuadro clínico avanzado del caso, no fue posible una intervención quirúrgica (7).
Las imágenes son indispensables para el diagnóstico, en edad pediátrica se prefieren imágenes multimodales, iniciando con ecografía abdominal, la cual es útil para determinar la presencia de neoplasia, pero no es específica para diagnóstico, por lo cual se debe realizar resonancia magnética nuclear (RMN) o tomografía computarizada (TAC) abdominal, para caracterizar tamaño, nódulos adyacentes y metástasis sistémica. Puede encontrarse imágenes grandes, heterogéneas, que contienen calcificaciones o cicatrices centrales (4).
Histológicamente, el CHC-FL está constituido por células tumorales grandes, poligonales, con citoplasma eosinofílico y abundante estroma fibroso que se encuentra alrededor de las células tumorales, características por las cuales, recibe el nombre fibrolamelar (8). Es poco frecuente la coexistencia de CHC y CHC-FL en el mismo paciente y cuando se encuentran, los dos componentes son claramente separados (9).
La resección quirúrgica es la piedra angular del tratamiento, pero en muchos casos la enfermedad es diagnosticada cuando el tumor compromete gran tamaño, por lo que la hepatectomía derecha es necesaria frecuentemente. La extensión de la cirugía depende de muchos factores como la localización, la proximidad a estructuras vasculares y ductales. Notablemente hasta el 85% del parénquima hepático puede ser removido, y se ha visto regeneración completa, incluso con quimioterapia postoperatoria (4).
A la paciente se le realizó hepatectomía derecha, ampliada teniendo en cuenta que el tumor ocupa los segmentos V, VI, VII y VIII, con lesión de 16 x 11 x 8,5 cm, de color verde amarillento, dejando bordes limpios y se dio alta hospitalaria para observación ambulatoria por parte de oncología y cirugía pediátrica.
El rol de terapia neoadyuvante es controversial, en muchos casos se ha evidenciado que la quimioterapia no tiene efecto en la supervivencia o recurrencia en paciente que ha recibido previamente tratamiento quirúrgico. Se estima una supervivencia general del 76% a los 5 años luego del tratamiento quirúrgico (10).
Conclusión
El diagnóstico es poco común en la edad pediátrica, además fue diagnosticado como hallazgo incidental, pues la paciente no presentaba sintomatología previa al trauma abdominal, que es la razón por la cual consulta, lo cual hace excepcional el caso reportado, a diferencia de lo reportado en la literatura no presenta hepatopatía crónica, lo que le confiere mejor pronóstico.
Referencias
- Lin C-C, Yang H-M. Fibrolamellar Carcinoma: A Concise Review. Arch Pathol Lab Med. 2018;142(9):1141–5.
- Graham RP. Fibrolamellar Carcinoma: What Is New and Why It Matters. Surg Pathol Clin. 2018;11(2):377–87.
- Kassahun WT. Contemporary management of fibrolamellar hepatocellular carcinoma: diagnosis, treatment, outcome, prognostic factors, and recent developments. World J Surg Oncol. 2016;14(1):151.
- Lim IIP, Farber BA, LaQuaglia MP. Advances in fibrolamellar hepatocellular carcinoma: a review. Eur J Pediatr Surg. 2014;24(6):461–6.
- Toro Rendón LG, García V, Peréz Cadavid JC, Hoyos Duque SI, Chávez Trujillo JF, Marín Zuluaga JI, et al. Hepatocarcinoma fibrolamelar un tumor de adultos jóvenes poco frecuente: Reporte de un caso. Rev Colomb Gastroenterol. 2014;29:433–8.
- Liu S, Chan KW, Wang B, Qiao L. Fibrolamellar Hepatocellular Carcinoma. Am J Gastroenterol. 2009;104(10):2617–24.
- Téllez-Zenteno JF, Hemández-Ronquillo L, Tapia-Rangel B, Garduño-Espinosa J. Hepatocarcinoma variedad fibrolamelar en una joven de 17 años de edad. Gac Med Mex. 1999;135(1):83–7.
- Ang CS, Kelley RK, Choti MA, Cosgrove DP, Chou JF, Klimstra D, et al. Clinicopathologic characteristics and survival outcomes of patients with fibrolamellar carcinoma: data from the fibrolamellar carcinoma consortium. Gastrointest Cancer Res. 2013;6(1):3–9.
- Stevens WR, Johnson CD, Stephens DH, Nagorney DM. Fibrolamellar hepatocellular carcinoma: stage at presentation and results of aggressive surgical management. AJR Am J Roentgenol. 1995;164(5):1153–8.
- El-Serag HB, Davila JA. Is fibrolamellar carcinoma different from hepatocellular carcinoma? A US population-based study. Hepatology. 2004;39(3):798–803.
|
