Introducción
El síndrome de Apert (SA) también llamado acrocefalosindactilia tipo I es un defecto genético dentro del grupo de las anomalías craneofaciales, específicamente aquellas que presentan craneosinostosis. Baumgartner hace las primeras menciones en 1842, seguido por Wheaton en 1894, sin embargo, la primera descripción completa de la enfermedad se le atribuye al doctor Eugene Apert en 1906.1 Se ha demostrado el efecto de la edad paterna avanzada en el desarrollo de la enfermedad, ya que en el 50% de los casos el padre tiene más de 35 años de edad.2 Se estima una prevalencia de 1:160.000 nacimientos, que es mayor en los asiáticos3 y los hispanos tienen el predominio más bajo (7.6 por millón de nacidos vivos4 y según la literatura revisada el SA representa cerca del 4.5% de todos los casos de craneosinostosis5.
El 40% de las craneosinostosis se diagnostica como parte de algún síndrome y de éstos el más común es el SA6. La etiología de este síndrome es la mutación del gen del Receptor 2 del factor del crecimiento fibroblástico (FGFR2), cuyo locus es 10q26. Se han identificado principalmente 2 mutaciones que causan la enfermedad, responsables del 99.2% de los casos de síndrome de Apert.7 Las dos involucran cambios en aminoácidos adyacentes a la región de unión del receptor. Ambas se manifiestan clínicamente igual, aunque la mutación S252W es más frecuente y se relaciona con paladar hendido y edad paterna avanzada, mientras que la mutación P253R se relaciona con sindactilia más severa en pies.8 Los hallazgos histológicos demuestran que la mutación en este receptor hace que se aumente el número de células precursoras que entran en la vía osteogénica, que termina con la osificación prematura de la calvaria, debido a la estimulación del receptor sin la unión de su ligando.9 La familia génica de los FGFRs (Specific Functions of Fibroblast Growth Factor Receptors) está compuesta por cuatro integrantes. El gen del FGFR1 está ubicado en el cromosoma 8, el del FGFR2 en el 10, FGFR3 en el 4 y FGFR4 en el cromosoma 5.10 Los diferentes FGFRs tienen una estructura proteica similar que consiste en tres dominios Ig (IgI, IgII y Ig III), un dominio transmembrana y dos dominios tirosin kinasa.11 La cascada intracelular de los FGFRs se ha relacionado con diversos procesos, entre ellos: mitogénesis, diferenciación, apoptosis y migración.12
El síndrome de Apert presenta una serie de signos clínicos diseminados a lo largo de toda la arquitectura corporal del individuo que lo padece, las diversas alteraciones y su frecuencia, como se muestran en la Tabla 1. Dadas la fuerte correlación clínica y la característica ósea distintiva, el método natural de detección es el ultrasonido. Las alteraciones anatómicas se detectan tan temprano como a las 17 semanas de gestación, lo que da oportunidad a una determinación mediante microensayo en sangre periférica e incluso se ha reportado interrupción del embarazo antes de la semana 20 de gestación.13 Las determinantes ultrasonográficas que llevan a la sospecha son la forma anormal de la cabeza en turribraquicéfalo (diámetro anteroposterior corto), nariz corta, y segundo y quinto dedos de manos y pies en sindactilia.14 El diagnóstico diferencial del síndrome de Apert se hace con otros síndromes que presentan craneosinostosis y alteración de las extremidades en grado variable como se muestran en la Tabla 2.
El tratamiento quirúrgico debe ser precoz, antes de los 6 meses de edad y va dirigido a descomprimir el espacio intracraneal, mejorar la función respiratoria y permitir el desarrollo normal de las distintas aéreas cerebrales. El tratamiento quirúrgico debe orientarse también a mejorar no solo el aspecto físico del niño, sino, además, las diversas alteraciones funcionales, sobre todo en cara y manos, aunque estas cirugías se realizan más tarde, alrededor de los 6 años.15
En la literatura se reporta una sobrevida no mayor a los 5 años, aunque existen pocos reportes de casos de paciente con este síndrome, por lo que consideramos importante reportar el caso de un paciente de 24 años de edad con SA.
Reporte de caso clínico
Masculino conocido desde los 2 años 2 meses de edad al servicio de Medicina Interna Pediatría para seguimiento y control. Dentro de sus antecedentes de importancia madre de 46 años de edad, es profesora, aparentemente sana. Padre finado de 50 años, por complicaciones de infarto miocardio, es hijo único. Producto de la gesta I, madre de 28 años, embarazo de 40 semanas de gestación curso con cuadros de infección de vías urinarias no recuerda cual. Obtenido por cesárea de urgencia por sufrimiento fetal, llora y respira al nacer, peso 3.6 kg talla 53 cm. APAGAR 8/8, sin complicaciones neonatales, dado de alta al binomio al cuarto día con diagnóstico de Síndrome de Apert. Es visto por dermatología, cirugía plástica, (craneofacial) y rehabilitación. Se ha intervenido quirúrgicamente en 13 ocasiones. No acude a la escuela, último grado escolar segundo año de bachillerato.
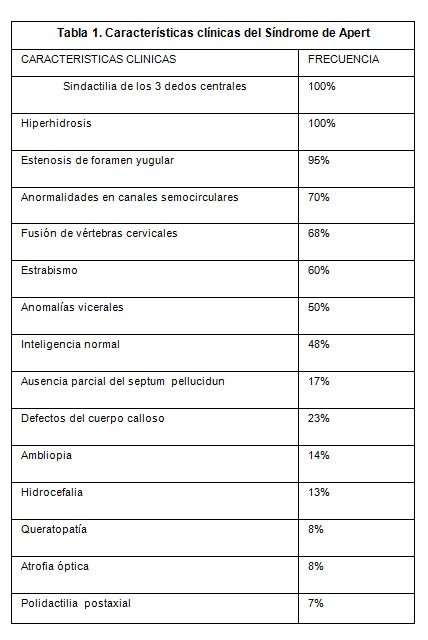
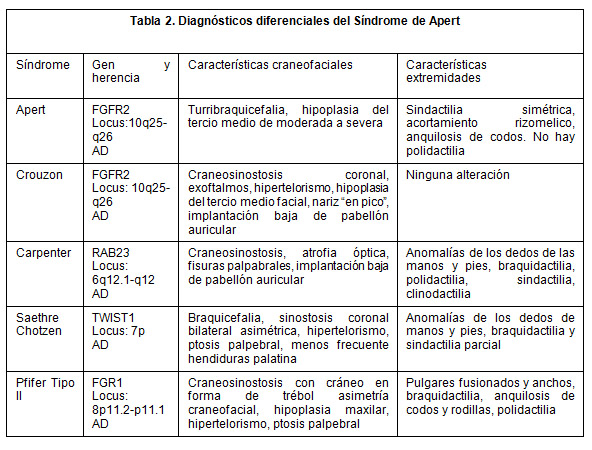
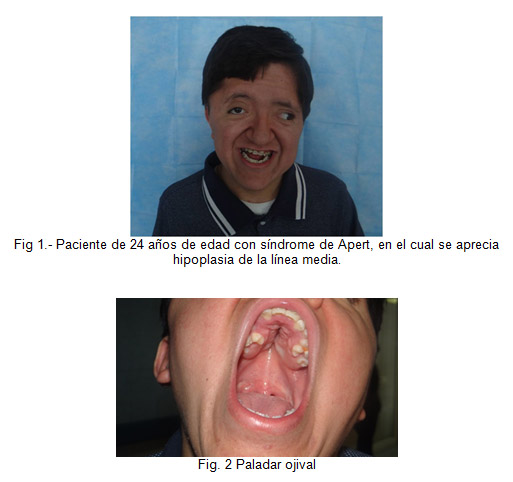
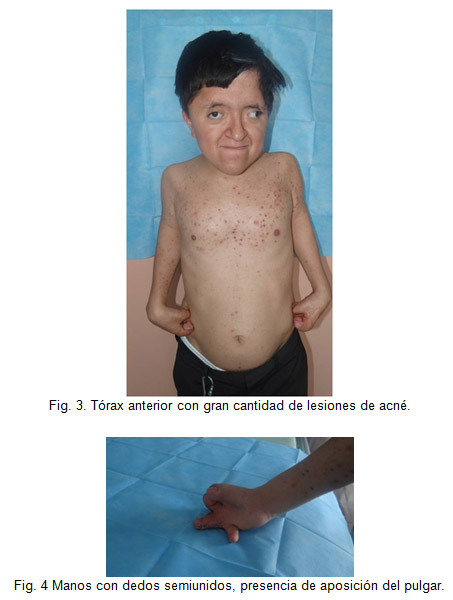
Exploración física con marcha claudicante de puntas, cabeza macrocefálico con placa en región temporal izquierda ligeramente móvil, con cabello implantado de acuerdo a la edad y género, hipoplasia de la línea media, frente amplia con exoftalmos, presencia de nistagmus (usa lentes), nariz en loro, narinas permeables, implantación baja de pabellones auriculares, faringe normal paladar ojival, piezas dentales normales, voz nasal, conductos permeables; cuello corto rígido, tráquea central movible, tórax corto ligeramente ancho con limitación en movimiento, de amplexión y amplexación, murmullo vesicular normal, espalada con gran cantidad de lesiones de acné, área cardiaca rítmica de buena intensidad sin soplos, abdomen plano blando depresible sin megalias, timpanismo y peristaltismo normal, con extremidades brazos corto con limitación de arcos de movilidad por anquilosis, de hombros y también de columna, codos en semiflexión y también muñecas con manos con dedos semiunidos, presencia de aposición del pulgar.
Se realiza estudio para el análisis de la mutación en el gen FGFR2 presentando un cambio en el estado heterocigoto de citocina por guanina en el nucleótido 1405 del gen, lo que condiciona un cambio de prolina por arginina en la posición 253 en la secuencia de aminoácidos del receptor (Pro253Arg).
El paciente fue sometido a 15 cirugías para la reparación de la craneosinostosis, sin ninguna complicación. Actualmente el paciente cuenta con 24 años de edad, sin retraso mental, con limitación parcial para realizar sus actividades secundario a las deformaciones físicas.
Discusión
El SA pertenece al grupo de las craneosinostosis por lo que se deben realizar diagnóstico diferencial con el síndrome de Cruzon, Chotzen, Pfeiffer, Noack, Carpenter y Goodman. El paciente demostró la triada clínica caracterizada por cráneo braquicéfalo, hipoplasia del tercio medio facial y sindactilia de manos y pies. Otras características faciales típicas son exoftalmo e implantación baja de pabellones auriculares, malformaciones, todas ellas, asociadas a sindactilia simétrica en los miembros superiores e inferiores de estos niños.
Por las alteraciones clínicas se sospechó del síndrome y se confirmó con la mutación genética. La fusión prematura de las suturas del cráneo en este síndrome da lugar a deformidades muchas veces asociadas con retardo mental y anomalías anatómicas y funcionales.
Conclusión
El diagnóstico generalmente se lo realiza en edades tempranas, debido a que se trata de un trastorno del desarrollo. Este se fundamenta en criterios clínicos y radiológicos. Su detección temprana es importante, ya que el tratamiento por parte de los diferentes especialistas debe comenzar desde el momento del nacimiento, para obtener de esta manera resultados adecuados desde el punto de vista funcional, estético y psicosocial. Generalmente requieren manejo multidisciplinario, el pronóstico es variable por lo que el consejo genético es importante para comprender la enfermedad, además de que se requiere más conocimiento sobre el manejo y posibles complicaciones en los pacientes con sobrevida mayor a la reportada en la literatura.
Referencia
- Apert ME. De l’acrocephalosyndactylie. Bull Mem Soc Med Hop Paris. 1906; 23: 1310-1330
- Tolarova MM, Harris JA, Ordway DE, Vargervik K. Birth prevalence, mutation rate, sex ratio, parents’ age, and ethnicity in Apert syndrome. Am J Med Genet. 1997; 72: 394-398.
- Cohen MM Jr, Kreiborg S, Lammer EJ, Cordero JF, Mastroiacovo P, Erickson JD, et al. Birth prevalence study of the Apert syndrome. Am J Med Genet. 1992; 42: 655-659
- Villarroel Goytia A, Hochtatter Arduz E. síndrome de apert. Gaceta boliviana 2007; 3: 60-2
- Cohen MM Jr, Kreiborg S, Lammer EJ, Cordero JF, Mastroiacovo P, Erickson JD, et al. Birth prevalence study of the Apert syndrome. Am J Med Genet. 1992; 42: 655-659
- Kan Sh, Elanko N, Johnson D, Cornejo-Roldan L, Cook J, Reich E et al. Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am J Hum Genet. 2002; 70: 472-486.
- S. Saritha, Sumangala, G. Supriya, M. Praveen Kumar. Apert syndrome (Acrocephalosyndactyly): a case report Int J Res Med Sci. 2013 Feb;1(1):36-40
- Cuttone JM, Brazis PT, Miller MT, Folk ER. Absence of the superior rectus muscle in Apert syndrome. J Pediatr Ophthalmol Strabismus. 1979; 16:349-54.
- Lomri A, Lemonnier J, Hott M, de Parseval N, Lajeunie E, Munich A, et al. Increased calvaria cell differentiation and bone matrix formation induced by fibroblast growth factor receptor 2 mutations in Apert syndrome. J Clin Invest. 1998; 101:1310-7.
- Muenke M, Wilkie AO. Craniosynostosis syndromes. In: Scriver CR, Beaudet AL, Valle D, Sly WS, Kinzler K, Vogelstein B, et al, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. 6117-46.
- Jockin YM, Katowitz JA, Fries PD, Hertle RW. Congenital craniofacial deformities: ophthalmologic considerations. In: Katowitz JA, editor. Pediatric oculoplastic surgery. New York: Springer-Verlag; 2002. p. 533-58.
- Mantilla-Capacho JM, Arnaud L, Diaz-Rodriguez M, Barros-Nunez P. Apert syndrome with preaxial polydactyly showing the typical mutation Ser252Trp in the FGFR2 gene. Genet Counsel. 2005; 16: 403-406.
- Ferreira J, Carter S, Bernstein P, Jabs E, Glickstein J, Marion R, Baergen R, Gross S. Second trimester molecular prenatal diagnosis of sporadic Apert syndrome following suspicious ultrasound findings. Ultrasound Obstet Gynecol. 1999; 14: 426-430.
- Stenirri S, Restagno G, Battista G, Alaimo G, Sbaiz L, Mari C, Genitori L, Maurizio F, Cremonesi L. Integrated strategy for fast and automated molecular characterization of genes involved in craniosynostosis. Clinical Chemistry. 2007; 53: 1767-1774.
- Reséndiz Martínez IA, Nava Uribe E. Síndrome de Apert. Acta médica grupo ángeles 2013; 11(4): 173-5
|
