El neuroblastoma es una neoplasia del sistema simpático, de origen embrionario, que proviene de las células de la cresta neural. Es el tumor extracraneal más frecuente en la edad pediátrica, se manifiesta principalmente en los primeros 5 años de vida. En cifras nacionales, corresponde a un 4.1% de los cánceres en niños, y es responsable de un 4.9% del total de la mortalidad infantil por cáncer (1). La ubicación del neuroblastoma suele ser retroperitoneal (65%), en relación a la médula suprarrenal o bien al tronco simpático en diferentes niveles (2).
La ubicación cervical es la menos frecuente, corresponde a un 2-5% del total de neuroblastomas (2), y suele tener un mejor pronóstico (3). La presentación clínica de estos pacientes es diversa, los síntomas más frecuentes son: masa palpable, disnea, estridor, síndrome de Horner (SdH) y disfagia (4–6). Se han descrito pocos casos en la literatura de neuroblastomas cervicales que se encuentren presentes desde el nacimiento; en estos casos, las expresiones clínicas más habituales son: masa cervical o SdH presentándose durante las primeras semanas o meses de vida (3,6–11).
A continuación, se presentan dos casos clínicos de pacientes con neuroblastoma cervical, tratados en el servicio de Cirugía Pediátrica del Hospital Roberto del Río, con distinta presentación clínica.
Caso clínico 1
Un paciente varón de 4 meses es derivado por aumento de volumen cervical izquierdo persistente desde el primer mes de vida. Inicialmente se sospechó un origen infeccioso de la masa, por lo que recibió tratamiento antibiótico, sin respuesta clínica. Al examen físico, la masa se describe como firme, lisa y bien delimitada, acompañada de ptosis palpebral ipsilateral, ya registrada por los padres en fotografías tomadas a la edad de 2 meses. También se constata miosis ipsilateral y heterocromía iridiana (Figura 1).
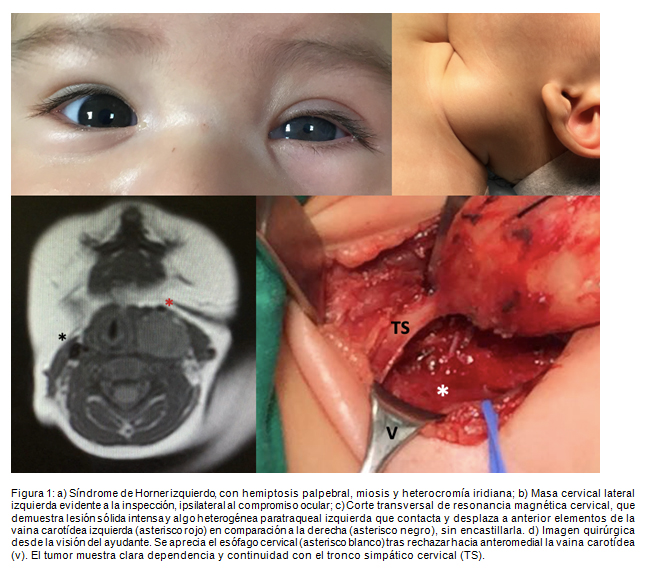
Se estudia inicialmente con una ecografía cervical que muestra una masa ovalada de 26mm de diámetro mayor, bien delimitada y heterogénea. Se complementa el estudio con una resonanciamagnética cervical que describe una lesión sólida de 22x16x30 mm, ubicada posterior a la arteria carótida y postero-medial al músculo esternocleidomastoideo izquierdo, que desplaza tráquea y esófago hacia la derecha sin colapsarlos, con señal hipointensa en T1 e hiperintensa en T2, con impregnación precoz en fase arterial.
Se sospecha neuroblastoma y se realizatumorectomía primaria. Se accede por cervicotomía izquierda sobre la lesión visible y palpable, que una vez disecada muestra clara relación con el tronco simpático a nivel paravertebral, y se reseca completamente. El paciente es extubado en pabellón evolucionando posteriormente sin dificultad respiratoria ni disfonía, y recibe un día de monitorización en unidad crítica.
La histopatología del tumor muestra un neuroblastoma pobremente diferenciado con histología favorable. El estudio de biología molecular revela n-myc no amplificado. El paciente es manejado con observación exclusiva. El seguimiento con cintigrama MIBG describe ausencia de tejido con actividad adrenérgica anormal, y en ecografías cervicales sucesivas no hubo recurrencia del tumor. A 2 años de la cirugía, el paciente se encuentra en buenas condiciones, con persistencia de SdH a izquierda, sin requerimiento de lágrimas artificiales.
Caso clínico 2
Un recién nacido de término de sexo masculino sin diagnósticos antenatales relevantes, presenta a la segunda hora de vida episodios de desaturación con requerimiento de oxígeno suplementario. Se estudia con radiografía de tórax y ecocardiografía, que descarta patología cardiopulmonar. Durante su segunda semana de vida se agrava agregándose al cuadro estridor y retracciones, por lo que se realiza una nasofibroscopía que evidencia un aumento de volumen paramediano derecho en la pared posterior de la faringe, correspondiente a una masa lisa, firme y bien delimitada, que desplaza, pero no compromete a la laringe (ver Figura 2). Se maneja la vía aérea con tubo orotraqueal y ventilación mecánica convencional. Al examen físico, el paciente no presenta masa cervical palpable ni SdH.
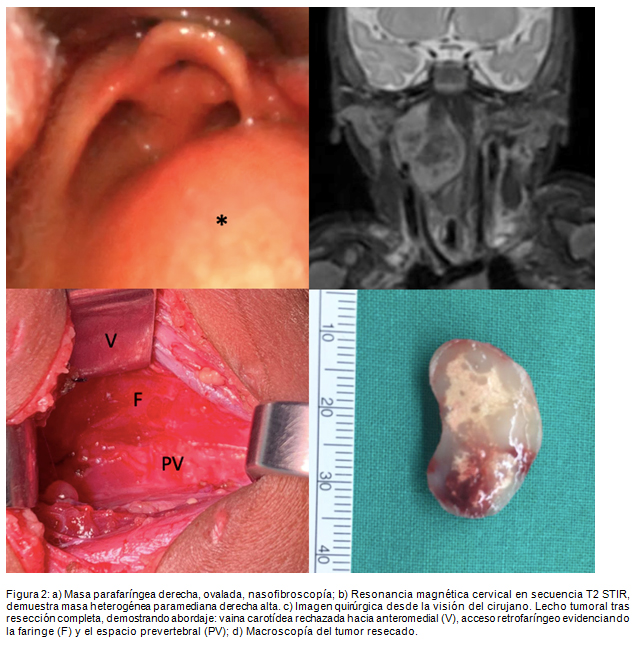
Se complementa el estudio con angiotomografía computada, encontrando una masa retrofaríngea de 28x28x16 mm, bien delimitada, que desplaza la vía aérea hacia la izquierda, sin realce significativo con contraste intravenoso, con hiperdensidades visibles en su interior, correspondientes a calcificaciones en casi su totalidad. Una resonancia magnética cervical, describe dicha lesión como heterogénea en secuencias T2/STIR, que restringe en secuencia de Difusión, y con moderado reforzamiento posterior al gadolinio.
El estudio sugiere la presencia de un neuroblastoma, por lo que se realiza una tumorectomía primaria. Se aborda por cervicotomía derecha, realizando extirpación completa de la masa, biopsia de ganglio cervical de aspecto patológico, sin necesidad de traqueostomía. El control inmediato postoperatorio con videolaringoscopía evidencia desaparición del efecto de masa. Se maneja inicialmente con glucocorticoides intravenosos e intubación orotraqueal que se retira al quinto día postoperatorio. El paciente evoluciona inicialmente con SdH derecho, conservando movilidad completa y simétrica de hombros.
La histopatología informa neuroblastoma poco diferenciado de histología favorable, y neoplasia neuroblástica en la adenopatía resecada. El estudio de biología molecular revela n-myc no amplificado. El estudio cintigráfico con metayodobencilguanidina (MIBG) concluye ausencia de tumor secundario ni remanente. El cintigrama ósea resulta sin lesiones óseas, y el mielograma, sin infiltración neoplásica. El paciente es clasificado como INSS 2B menor de 1 año y sin amplificación n-myc, siendo susceptible de observación postoperatoria conforme protocolo local (PINDA). A 5 meses de la cirugía, el paciente se encuentra asintomático, sin evidencia de ptosis palpebral derecha ni anisocoria. Se alimenta sin dificultad. Al control ecográfico se encuentra libre de recidiva.
Discusión
Las masas cervicales en neonatos y lactantes suelen relacionarse con alteraciones del desarrollo, como por ejemplo el quiste tirogloso, remanentes de arcos faríngeos, y malformaciones vasculares. También destacan masas adquiridas de carácter inflamatorio, como adenopatías (12), mientras que neoplasias como el neuroblastoma no suelen ser sospechadas en primera instancia. El SdH por su parte, refleja una disfunción de la vía óculosimpática que se manifiesta con ptosis palpebral, miosis y en algunos casos con heterocromía iridiana y anhidrosis ipsilateral. Lo más frecuente es que sea producto de trauma o tumores que compriman o se originen en la vía óculosimpática, como ocurre con el neuroblastoma cervical. Se ha reportado SdH en niños con neuroblastomas en otros sitios y en ausencia de masa cervical (13), lo que podría explicarse por efecto paraneoplásico, disgenesia del sistema simpático, o por regresión espontánea y falta de detección del tumor en imágenes (8).
La obstrucción de vía aérea superior eventualmente puede darse en contexto de masas de carácter malformativo (quistes laríngeos saculares, malformación vascular linfática), o eventualmente neoplásico (hemangioma subglótico, teratomas). Se trata de una presentación atípica del neuroblastoma cervical (14).
Actualmente el neuroblastoma se clasifica de acuerdo al compromiso local y diseminación según el International Neuroblastoma Risk Group Staging System (INRGSS), un sistema de etapificación preoperatoria en base a imágenes. Se divide a los Neuroblastomas en (15): L1 (localizados, sin factores de riesgo imagenológicos, “IDRF”); L2 (localizados, con IDRF); M (presencia de metástasis); y SM (enfermedad metastásica en menores de 18 meses, limitadas a la piel, hígado y/o médula ósea). La presencia o ausencia de IDRF ha demostrado tener implicancias en la complejidad quirúrgica, tiempo hospitalario y morbilidad perioperatoria (16), aunque su impacto en la sobrevida se ha cuestionado en algunas series (17). En posición cervical, los IDRF son: 1) encastillamiento de vasos mayores (carótida, vertebral, yugular interna); 2) extensión a base de cráneo; 3) compresión traqueal; y 4) extensión cervicotoráxica (15). Ninguno de nuestros pacientes presentó tales factores, clasificándose como L1 y siendo susceptibles de resección primaria sin tratamiento neoadyuvante.
El manejo específico del neuroblastoma depende de la clasificación de riesgo del paciente, que considera, además de la etapificación clínico-imagenológica, la edad al diagnóstico y características patológicas y biológicas tales como la clasificación histológica, grado de diferenciación, la presencia de amplificación de n-myc, ploidías del DNA y alteraciones cromosómicas segmentarias (18).
Existen diferentes alternativas de tratamiento para el neuroblastoma, que van desde el seguimiento expectante en casos seleccionados (19), solo quirúrgico, o terapias complementarias como quimioterapia, radioterapia, terapias biológicas y trasplante de precursores hematopoyéticos. El pronóstico suele ser mejor en neuroblastomas extraadrenales, y cabe destacar que en tumores clasificados como bajo riesgo la sobrevida a 5 años supera el 85% (15).
La literatura respecto al tratamiento quirúrgico de neuroblastomas cervicales es reducida. Un estudio realizado por la Sociedad Americana de Oncología en una cohorte de 915 pacientes con diagnóstico de neuroblastoma de bajo riesgo (20), señala que la terapia quirúrgica exclusiva puede ser curativa en la mayoría de los pacientes de ese grupo, y el uso de quimioterapia debe ser restringido a casos específicos. Sin embargo, en este estudio no se señala el número de casos de neuroblastomas cervicales ni sus resultados específicos. Otra revisión norteamericana de 220 casos, que incluye menos de 10 casos de neuroblastoma cervical, estudia el impacto de la extensión de la resección en el control local y la supervivencia de pacientes con neuroblastomas de alto riesgo (21). Este estudio llega a la conclusión de que la resección de un 90% o más del tumor mejora significativamente la sobrevida libre de eventos, y disminuye la incidencia acumulada de progresión local. Una serie egipcia de 202 pacientes (22), de los cuales 10 tenían neuroblastoma cervical tratados con quimioterapia neoadyuvante, evalúa el rol de la cirugía y el tamaño de la resección de neuroblastomas inicialmente irresecables no metastásicos. Este estudio concluye que la resección quirúrgica fue el único factor de riesgo significativo respecto a la sobrevida.
Para la resección de neuroblastomas cervicales se han descrito accesos transorales, transcervicales, y transhioideofaringotomía (6), que depende de la localización y características del neuroblastoma. En los dos casos presentados, el abordaje fue por cervicotomía transversa lateral accediendo al espacio retrofaríngeo o prevertebral por lateral y posterior a la vaina carotídea, un plano mayormente avascular, buscando reducir el riesgo de lesión vasculonerviosa. Tomando en cuenta el tamaño reducido del cuello de estos pacientes, es importante conocer la anatomía cervical para anticipar las estructuras nerviosas vecinas evitando su lesión directa o tracción excesiva.
Hay evidencia de regresión espontánea del neuroblastoma, lo que justifica un manejo expectante en casos seleccionados. Existen a la fecha criterios bien definidos y consensuados para la observación en pacientes con diagnóstico de neuroblastoma suprarrenal (19). La observación en enfermedad extra adrenal ha tenido resultados promisorios en grupos europeos y orientales (23,24), y existe el protocolo ANBL1232, realizado por un grupo norteamericano, que estudia el manejo expectante en neuroblastomas de bajo riesgo, que aún no ha sido publicado (25). Un estudio alemán en el que se siguieron 358 pacientes con neuroblastoma, de 15 pacientes que tenían neuroblastoma cervical, 11 de ellos regresaron espontáneamente (23). En general, se consideran para observación exclusiva aquellos pacientes sin síntomas de riesgo vital, que en el contexto de neuroblastoma cervical involucra el compromiso respiratorio y cardiovascular. Nuestro primer caso presentado (cuya clínica tan solo consistía en SdH y masa cervical, sin riesgo vital) podría ser considerado dentro de este tipo de manejo, aunque no fue planteado en esta ocasión por decisión del equipo tratante; se ofreció la cirugía, que resultó sin morbilidad significativa y fue curativa para el niño. El segundo caso, que se presentó con obstrucción de vía aérea superior requiriendo asistencia ventilatoria, no era susceptible de manejo expectante.
Conclusión
Un adecuado manejo del neuroblastoma cervical requiere de un diagnóstico precoz, para lograr un tratamiento óptimo y con una baja morbilidad. Es por esta razón, que una masa cervical en un neonato, o bien el hallazgo de SdH sin causa traumática que lo explique, deberían hacernos sospechar un neuroblastoma, y buscarlo dirigidamente. También debe considerarse ese diagnóstico en presencia de compromiso obstructivo de vía aérea superior en contexto de una masa cervical sólida probada por imágenes. Tras un estudio imagenológico acabado, la cirugía puede jugar un rol fundamental en su tratamiento y ser curativa para algunos pacientes. Un grupo bien definido de niños sin riesgo vital también podría ser susceptible de observación hasta regresión espontánea.
Agradecimientos
Se extiende un agradecimiento especial a los colegas Margarita Aldunate, Benigno Montenegro y Néstor Quitral, y a las unidades de Neonatología del Hospital San José, y Radiología, Otorrinolaringología y Oncología del Hospital Roberto del Río.
Referencias
- Vallebuona C. Primer Informe del Registro Nacional de Cáncer Infantil de Chile (Menores de 15 Años), RENCI, Quinquenio 2007 - 2011. Primera Edición. Departamento de Epidemiología Ministerio de Salud; 2018: 56-63.
- Vo KT, Matthay KK, Neuhaus J, London WB, Hero B, Ambros PF, et al. Clinical, Biologic, and Prognostic Differences on the Basis of Primary Tumor Site in Neuroblastoma: A Report From the International Neuroblastoma Risk Group Project. J Clin Oncol. 2014 Oct;32(28):3169–76.
- Isaacs H. Fetal and Neonatal Neuroblastoma: Retrospective Review of 271 cases. Fetal Pediatr Pathol. 2007 Jan;26(4):177–84.
- Yokoya S, Suda T, Koyama M, Yanagisawa M, Kamoshita S, Miyakawa K, et al. Retropharyngeal neuroblastoma causing airway obstruction in a newborn—Survival with surgical treatment alone. J Pediatr Surg. 1982 Apr;17(2):180–1.
- Alvi S, Karadaghy O, Manalang M, Weatherly R. Clinical manifestations of neuroblastoma with head and neck involvement in children. Int J Pediatr Otorhinolaryngol. 2017 Jun;97:157–62.
- Okazaki T, Ohshita M, Furukawa M, Ikeda K, Ozaki Y, Lane GJ, et al. Retropharyngeal neuroblastoma in a neonate: case report and literature review. Pediatr Surg Int. 2007 Sep;23(10):1023–6.
- Cardesa-Salzmann TM, Mora-Graupera J, Claret G, Agut T. Congenital cervical neuroblastoma. Pediatr Blood Cancer. 2004 Dec;43(7):785–7.
- Zafeiriou DI, Economou M, Koliouskas D, Triantafyllou P, Kardaras P, Gombakis N. Congenital Horner’s syndrome associated with cervical neuroblastoma. Eur J Paediatr Neurol. 2006 Mar;10(2):90–2.
- Jackson JR, Tran HC, Stein JE, Shimada H, Patel AM, Marachelian A, et al. The clinical management and outcomes of cervical neuroblastic tumors. J Surg Res. 2016 Jul;204(1):109–13.
- Nellis JC, Rivas A, Brown DJ, Ishman SL. Neonatal neck mass with associated Horner Syndrome: A unique presentation of neuroblastoma. Int J Pediatr Otorhinolaryngol Extra. 2013 Sep;8(3):79–81.
- White JB, Meany HJ, Ahmed AA, Preciado D. Neuroblastoma: An unusual diagnosis for a pediatric retropharyngeal mass. Int J Pediatr Otorhinolaryngol Extra. 2011 Mar;6(2):62–4.
- Meier J, Grimmer J. Evaluation and Management of Neck Masses in Children - American Family Physician. Am Acad Fam Physicians. 2014 Mar;89(5):353-358.
- Gibbs J, Appleton RE, Martin J, Findlay G. Congenital Horner Syndrome Associated with Non-cervical Neuroblastoma. Dev Med Child Neurol. 2008 Nov 12;34(7):642–4.
- Bhatt J, Prager JD. Neonatal Stridor. Clin Perinatol. 2018 Dec;45(4):817–31.
- Newman EA, Abdessalam S, Aldrink JH, Austin M, Heaton TE, Bruny J, et al. Update on neuroblastoma. J Pediatr Surg. 2019 Mar;54(3):383–9.
- Monclair T, Brodeur GM, Ambros PF, Brisse HJ, Cecchetto G, Holmes K, et al. The International Neuroblastoma Risk Group (INRG) Staging System: An INRG Task Force Report. J Clin Oncol. 2009 Jan 10;27(2):298–303.
- Phelps HM, Ndolo JM, Van Arendonk KJ, Chen H, Dietrich HL, Watson KD, et al. Association between image-defined risk factors and neuroblastoma outcomes. J Pediatr Surg. 2019 Jun;54(6):1184–91.
- Cohn SL, Pearson ADJ, London WB, Monclair T, Ambros PF, Brodeur GM, et al. The International Neuroblastoma Risk Group (INRG) Classification System: An INRG Task Force Report. J Clin Oncol. 2009 Jan 10;27(2):289–97.
- Nuchtern JG, London WB, Barnewolt CE, Naranjo A, McGrady PW, Geiger JD, et al. A Prospective Study of Expectant Observation as Primary Therapy for Neuroblastoma in Young Infants: A Childrenʼs Oncology Group Study. Ann Surg. 2012 Oct;256(4):573–80.
- Strother DR, London WB, Schmidt ML, Brodeur GM, Shimada H, Thorner P, et al. Outcome After Surgery Alone or With Restricted Use of Chemotherapy for Patients With Low-Risk Neuroblastoma: Results of Children’s Oncology Group Study P9641. J Clin Oncol. 2012 May 20;30(15):1842–8.
- von Allmen D, Davidoff AM, London WB, Van Ryn C, Haas-Kogan DA, Kreissman SG, et al. Impact of Extent of Resection on Local Control and Survival in Patients From the COG A3973 Study With High-Risk Neuroblastoma. J Clin Oncol. 2017 Jan 10;35(2):208–16.
- Ahmed G, Fawzy M, Elmenawi S, Elzomor H, Yosif Y, Elkinaai N, et al. Role of surgery in localized initially unresectable neuroblastoma. J Pediatr Urol. 2018 Jun;14(3):231–6.
- Hero B, Simon T, Spitz R, Ernestus K, Gnekow AK, Scheel-Walter H-G, et al. Localized Infant Neuroblastomas Often Show Spontaneous Regression: Results of the Prospective Trials NB95-S and NB97. J Clin Oncol. 2008 Mar 20;26(9):1504–10.
- Arakawa A, Oguma E, Aihara T, Kishimoto H, Kikuchi A, Hanada R, et al. Long-Term Follow-Up Results of the Observation Program for Neuroblastoma Detected at 6-Month Mass Screening. J Pediatr. 2014 Oct;165(4):855-857.
- Response and Biology-Based Risk Factor-Guided Therapy in Treating Younger Patients With Non-high Risk Neuroblastoma - Tabular View - ClinicalTrials.gov [Internet]. [cited 2020 May 7]. Available from: https://clinicaltrials.gov/ct2/show/record/NCT02176967
|
