Introducción
La violencia
intrafamiliar se refiere a todas las formas
de abuso que ocurren en las relaciones entre
los miembros de una familia. Esto incluye
las conductas que, por acción u omisión,
dañen física o psicológicamente
a otro miembro de la familia. Dentro de estas
expresiones de violencia se encuentra el maltrato
infantil. El maltrato infantil se puede expresar
como abuso físico, que es cualquier
acción no accidental por parte de los
cuidadores o padres, que provoque daño
físico o sea causal de enfermedad en
los niños. La intensidad puede variar
desde una contusión leve hasta una
lesión mortal. Éste puede ser
también pasivo, como el abandono físico,
en el que las necesidades básicas no
son atendidas temporal o permanentemente.
A continuación
mostraremos un caso clínico, que a
la luz de los antecedentes y de una historia
poco clara llevó a pensar a los pediatras
en un caso de maltrato infantil, pero que
el posterior estudio reveló otros diagnósticos.
Caso
clínico
IRZ, paciente
con fecha de nacimiento 23 noviembre 2002.
La madre consulta el 17 de enero de 2004 por
la siguiente historia: hace 3 semanas presenta
vómitos ocasionales por dos días,
que ceden solos. Hace 2 semanas, estando con
sus abuelos, se golpea en la zona frontal
derecha sin pérdida de conciencia.
No consulta y sólo presenta un leve
hematoma. Dos días después se
golpea nuevamente sin pérdida de conciencia.
Hace 2 semanas presenta secreción en
los ojos. Consulta médico particular
y se diagnostica conjuntivitis. Este cuadro
persiste a pesar del tratamiento. El motivo
actual de consulta fue la aparición
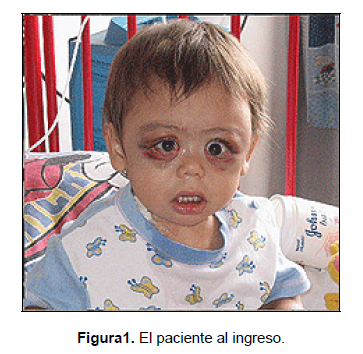
de equimosis bipalpebral desde el amanecer,
que la madre no logra explicar (Figura 1).
Al examen físico
el niño se observa hidratado, activo,
pero irritable. Presenta edema facial en ambas
órbitas con aumento de volumen bilateral
en región temporal, hemorragia subconjuntival
bilateral, equimosis periorbitaria, hematoma
en ángulo externo del ojo derecho,
exoftalmos bilateral y estrabismo convergente.
Se observa pálido, pero con buen llene
capilar. Presenta trastorno de la marcha,
irritabilidad y diarrea.
Se realizan
radiografías que muestran fracturas
antiguas.
Los diagnósticos
son:
- Fractura antigua de fémur
bilateral
- Fractura antigua de
radio izquierda
- Fractura antigua de
humero derecha
- Hemorragia subconjuntival
- Maltrato Infantil
El paciente
es hospitalizado por la sospecha de maltrato
infantil. Una vez ingresado, se realiza Tomografía
axial computada (TAC) de cerebro y órbita
sin contraste, la que muestra imagen hiperdensa
de bordes irregulares radiados que comprometen
cuerpo y alas mayores del esfenoides, llegando
a paredes laterales de las órbitas,
desplazando globo ocular en ambos lados.
A derecha existe
compromiso fronto-basal comprimiendo tejido
cerebral. Hacia la región occipital
izquierda existe imagen pequeña hiperdensa
leve extradural y otra que rodea la zona del
peñasco izquierdo.
Se diagnostica
tumor de base de cráneo y maltrato
infantil.
Se realizaron
exámenes que se describen a continuación:
- Hemograma: hemoglobina
7.8 mg/dl, leucocitos 10.200, plaquetas
406.000
- VHS: 31 mm/hora
- Perfil bioquímico:
normal, excepto por leve aumento de transaminasas
- Pruebas de coagulación:
normal
- SPOT test: intensamente
positivo en 2 muestras separadas por 24
hrs.
- Mielograma: celularidad
normal, 90% de células mononucleares
con escaso citoplasma, sin nucleolo, cromatina
laxa, algunos en acúmulos celulares,
compatible con infiltración de células
de neuroblastoma.
- Cariograma de médula
ósea: no se obtienen mitosis
- Ecografía abdominal:
discreta esplenomegalia y masa en región
suprarrenal izquierda de 4 x 5 cm. con microcalcificaciones
- Radiografía de
tórax: derrame pleural escaso, sin
sombras patológicas.
- TAC de abdomen: masa
suprarrenal izquierda que se extiende por
retroperitoneo posterior izquierdo compatible
con neuroblastoma
- TAC cerebral: imágenes
extradurales que captan contraste a nivel
de la base de cráneo y de las órbitas
- Cintigrafía ósea
trifásica con tecnesio 99 –
MDP: demuestra captación aumentada
e irregular en calota y en ambas regiones
periorbitarias, además de ambos fémures
y tibias
- Tirosin hidroxilasa:
positivo
- Amplificación
N-myc: negativo en médula ósea
- No se realiza metayodobenzilguanidina
(MIBG)
Con estos exámenes
se diagnostica Neuroblastoma en etapa IV.
Marco
teórico
El neuroblastoma
se origina de las células primordiales
de la cresta neural que durante la embriogénesis
migran para formar la glándula suprarrenal
y los ganglios simpáticos del sistema
nervioso simpático. Pueden presentarse
en cualquier sitio del sistema nervioso simpático,
con inclusión de cerebro, cuello, mediastino,
pelvis ganglios simpáticos paraaórticos
y glándula suprarrenal.
Epidemiología
El neuroblastoma
representa el 8,7% de todos los tumores pediátricos,
siendo el segundo tumor sólido más
frecuente en la niñez (2). Tiene una
incidencia anual de 8,7 casos por 1.000.000
niños menores de 15 años (5),
siendo similar a la de otros países
industrializados.
La edad media
al diagnóstico es de 22 meses. Al diagnóstico,
el 25% es menor de un año; 50% menor
de dos años; 75% menor de cuatro años
y 90% menor de 10 años.
Se ha relacionado
el neuroblastoma con otras alteraciones de
las células de la cresta neural como:
agangliosis de colon y el síndrome
de hipoventilación central.
No se ha descrito
asociación clara entre ningún
factor ambiental, químico, o biológico
y el desarrollo de neuroblastoma. Existe un
grupo de pacientes con una predisposición
familiar a padecer neuroblastoma que sigue
un patrón autonómico dominante
(2).
Patología
e histología
Entre las 17
y 20 semanas de gestación se desarrollan
nódulos de neuroblastoma en el feto
sano y regresan hacia el término del
embarazo o poco después de nacer. La
citogenética del tumor se caracteriza
por células con deleción del
brazo corto del cromosoma 1, presencia de
cuerpos cromatínicos dobles diminutos
(dmins) y regiones que se tiñen homogéneamente
(HSR) (2). Microscópicamente, el neuroblastoma
típico consiste en células uniformes
pequeñas compuestas por un denso núcleo
hipercromático y muy escaso citoplasma,
con presencia de neuropilos o procesos neuríticos
característicos.
Marcadores
celulares del Neuroblastoma
Genes TRK y
receptores neurotróficos
Ciertos marcadores
celulares en adición a la diferenciación
celular neuronal tienen importancia pronóstica
en el neuroblastoma, debido a que su malignidad
puede representar una falla en el estímulo
o en la célula para iniciar la diferenciación
celular. Estos marcadores son péptidos
neurotróficos e incluyen el Factor
de crecimiento neuronal (NFG), el Factor neurotrófico
derivado del cerebro (BDNF), la Neurotrofina-3
(NT-3) y la Neurotrofina-4/5 (NT-4/5). Cada
uno de ellos tiene un receptor de alta afinidad
específico (NFG ? TRK-A, BDNF y NT-4/5
? TRK-B, NT-3 ? TRK-C). Todos se unen a receptores
de baja afinidad para el factor de crecimiento
neuronal (LNGFR)
NFG: La expresión de TRK-A mRNA se
relaciona con ausencia de amplificación
del gen MYCN (se asocia a estadios tempranos
de la enfermedad con un pronóstico
más favorable). La disminución
de NFG produce muerte celular en estos tumores,
sugiriendo sensibilidad a la diferenciación
o regresión como resultado de la apoptosis.
La expresión de TRK-A y la amplificación
de MYCN son dos factores poderosos en el pronóstico
del neuroblatoma.
La expresión de LNGFR se asocia a un
mejor pronóstico. La mejor supervivencia
se ve en la asociación de TRK-A y LNGFR
mRNA, por regresión espontánea
del tumor.
BDNF: TRK-B se expresa exclusivamente en neuroblastomas
agresivos con amplificación de MYCN.
BDNF promovería la supervivencia celular
e induciría crecimiento neurítico
e impediría la acumulación de
vinblastina en las células tumorales
(posible efecto de resistencia a la quimioterapia).
Gen TRK: tiene gran importancia en la diferenciación
y regresión del neuroblastoma. Se vincula
con una mejoría significativa en la
supervivencia que en aquellos tumores que
no expresan el receptor. La supervivencia
a dos años con tres factores favorables
(TRK, ausencia de amplificación de
MYCN e histología) es de 90%, frente
al 0% de los pacientes con estos tres factores
negativos.
Antígeno nuclear de proliferación
celular (PNCA)
Es una subunidad de la DNA polimerasa. Los
neuroblastomas de mayor malignidad se caracterizan
por poseer elevadas tasas de PCNA. Se le ha
visto asociado en los factores infecciosos
del desarrollo del neuroblastoma.
Marcadores
séricos del neuroblastoma
Metabolitos
de catecolaminas
El ácido
homovanílico (HVA) es el mayor metabolito
de la DOPA y la dopamina, mientras que el
ácido vanilmandélico (VMA) es
el mayor metabolito de la noradrenalina y
la adrenalina. Concentraciones elevadas de
ellos en la orina son marcadores importantes
en la progresión en neuroblastomas
sin amplificación de MYCN, y sus valores
séricos sirven como indicadores pronósticos.
La sensibilidad para detectar un neuroblastoma
utilizando VMA y HVA es de 96% usando cromatografía
líquida de alta definición.
Ferritina
El grado de
aumento de ferritina sérica se relaciona
con el estadio de la enfermedad. Su incremento
puede ser signo de crecimiento tumoral, de
producción de ferritina por las células
tumorales, o incremento de ferritina-hierro
que puede potenciar el crecimiento tumoral.
Se identifican tres grupos distintos con diferente
pronóstico:
- Ferritina normal y edad
menor de dos años: supervivencia
a los dos años de 93%
- Ferritina normal y edad
mayor de dos años: supervivencia
a los dos años de 38%
- Ferritina elevada: supervivencia
a los dos años de 19%
Enolasa
neurona específica (NSE)
Es una proteína
citoplasmática de las células
neurales. Está elevada en el 96% de
los sujetos con metástasis al diagnóstico.
Sus niveles aumentan con el estadio de la
enfermedad (mayor a 100 ng/ml), lo que se
asocia a menores tasas de superviviencia (14%
a los 30 meses).
Gangliósido
GD2
Se expresa de
manera predominante en el neuroblastoma, detectándose
fácilmente en el plasma. Es un modulador
de la interacción tumor-huésped.
Se ha informado su disminución como
respuesta al tratamiento y un incremento durante
la recurrencia del paciente.
Cifras aumentadas al momento del diagnóstico
se asocia a mayor severidad y menor supervivencia
en estadios avanzados.
Deshidrogenada
láctica (LDH)
Marcador no
específico del neuroblastoma. Su aumento
se relaciona con el rápido crecimiento
celular o con tumores grandes. Concentraciones
mayores a 1500 U/ml se relacionan con un mal
pronóstico. Se utiliza como un marcador
de la actividad de la enfermedad o respuesta
al tratamiento.
Manifestaciones
clínicas
El
neuroblastoma se presenta como una masa tumoral
a lo largo de las vías neurales simpáticas.
El sitio del tumor primario es tan diverso
como la localización del tejido del
sistema nervioso autónomo y varía
según la edad del paciente.
Las manifestaciones
por segmento corporal se resumen en la tabla
1. La presentación clínica
más común es una masa abdominal
asintomática dura y fija (sobre todo
si hay diseminación hepática).
El neuroblastoma torácico es, por lo
general, un hallazgo radiográfico,
y raramente ocasiona síndrome de la
vena cava superior (puede manifestarse como
dificultad respiratoria).

En la pelvis
puede producir alteraciones vesicales o intestinales,
por lo general debidas a la compresión
de estos órganos por la masa. Es rara
la obstrucción intestinal.
En 60 a 65%
existe enfermedad metastásica al hacer
el diagnóstico, que se caracteriza
por presentar: pérdida de peso, anorexia,
compromiso del estado general y fiebre.
Otros signos
y síntomas son astenia, adinamia, irritabilidad,
letargia, dolor óseo (si hay diseminación
ósea), adenopatías, equimosis
peripalpebral, movimientos incontrolables
del ojo, parálisis de extremidades
inferiores y movimiento descoordinado.
También
se pueden presentar signos y síntomas
de excesiva secreción de catecolaminas
como diaforesis, palidez, hipertensión,
falla del crecimiento, cefalea, rubor facial,
palpitaciones, distensión abdominal,
diarrea acuosa de difícil tratamiento
e hipopotasemia.
Métodos
diagnósticos
Para el diagnóstico
hace falta cumplir uno de estos dos criterios:
- El diagnóstico
anatomopatológico del tumor con o
sin aumento de la excreción de catecolaminas
o sus metabolitos en la orina.
- Mielograma (mínimo
dos) de ambas espinas iliacas o un aspirado
o biopsia de médula ósea positiva
para células con positividad simultánea
de las catecolaminas o metabolitos en orina
o suero.
Desde el punto de vista inmunocitoquímico
se recomienda la utilización de, al
menos dos anticuerpos monoclonales.
Estudios
de laboratorio
Más
del 90% de los pacientes excretan niveles
altos de catecolaminas y sus metabolitos en
orina (HVA, AVM) debido a que las células
tumorales carecen de metiltransferasa, una
de las enzimas que metabolizan la dopamina.
Para que los valores en orina de 24 horas
sean positivos, tienen que ser superiores
a 2.5 desviaciones estándar respecto
a valores normales según la edad del
niño. Si las catecolaminas son negativas,
debe determinarse la dopamina sérica
o urinaria para identificar tumores indiferenciados.
Por último, el 10% de los neuroblastomas
no produce catecolaminas.
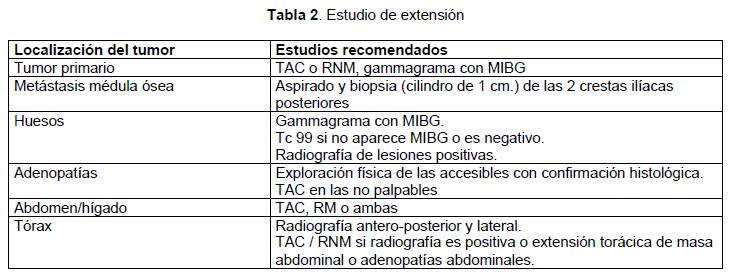
Estudios
de imágenes
Se recomiendan
tanto para el diagnóstico como para
definir la extensión tumoral (Tabla
2 y tabla
3).

Radiografía
simple
Es útil
para el diagnóstico del neuroblastoma
toráxico. Se identifica una imagen
radioopaca homogénea, con bordes bien
definidos en el mediastino posterior, con
o sin calcificaciones en su interior. La erosión
cortical costal es otro hallazgo que orienta
al diagnóstico. La radiografía
simple también permite descartar otras
causas de captación gamma aumentada
en huesos afectos.
Ultrasonido
Es esencial
en la atención inicial de pacientes
con masa abdominal. Permite definir su localización
y delimitar los vasos y los órganos
infiltrados por el tumor. El ultrasonido de
color doppler proporciona información
de la vascularidad intratumoral.
Tomografía
computada
Es imprescindible
para definir los límites del tumor,
su volumen y relación con las estructuras
vecinas (Figura 2); extensión hacia
el interior del conducto medular, los ganglios
linfáticos y la presencia de enfermedad
metastásica pleuropulmonar, abdominal
o de cabeza y cuello. La TAC helicoidal es
útil para definir la relación
del tumor con los grandes vasos.
Resonancia
nuclear magnética (RNM)
Tiene una sensibilidad
y especificidad para el diagnóstico
de 100%. Define mejor la anatomía vascular
y las metástasis a médula ósea.
También ofrece mayor precisión
para evaluar la enfermedad metastásica
y la extensión intraespinal del tumor
y a ganglios linfáticos.
Gammagrama
con metayodobenzilguanidina
La MIBG es transportada
y almacenada en gránulos de las células
cromafines al igual que la noradrenalina.
Es captada por la mayoría de los neuroblastomas,
pero no por el tejido óseo normal,
lo que la hace muy útil en la detección
del tumor primario y de las metástasis.
En caso que el tumor no capte MIBG (menos
del 5%) debe realizarse gammagrafía
con Tc 99.
Diagnóstico
clínico
Existen situaciones
en las que la obtención de material
para estudio histológico es de alto
riesgo vital para el paciente (etapa 4S);
en estos casos está permitido hacer
el diagnóstico de neuroblastoma, siempre
que se cumplan las siguientes condiciones:
- Tumor altamente sugerente
de neuroblastoma por localización,
con una ecogenicidad heterogénea
y/o calcificaciones.
- Presencia de tumor típico
en médula ósea
- Catecolaminas elevadas
en suero y orina
- Captación cintigráfica
evidente de MIBG en territorio tumoral.
Diagnóstico
diferencial
En ocasiones
resulta difícil distinguir el neuroblastoma
de tumores benignos, como el ganglioneuroma,
feocromocitoma y otros tumores neurales, que
también pueden secretar catecolaminas
y captar MIBG. Frente a estos casos, los datos
de genética molecular son definitivos,
por lo que es fundamental tener suficiente
material procedente del tumor.
La distinción
del neuroblastoma con las otras causas de
masa abdominal, debe basarse en la edad del
paciente y la clínica. Así,
por ejemplo, si la masa es retroperitoneal
no renal en un neonato, el teratoma y la hemorragia
suprarrenal deberán tenerse también
presentes. En los niños más
grandes se debe tener presente el tumor de
Willms y los tumores hepáticos siendo
el más frecuente el hemangioendotelioma.
Dentro de los tumores
del mediastino posterior en el niño,
aproximadamente 95% de ellos corresponde a
tumores de origen neurogénico y dentro
de estos, el neuroblastoma es el más
frecuente. Otros tumores menos comunes incluyen
ganglioneuroma, ganglioneuroblastoma, neurofibroma.
Las lesiones metastásicas
plantean un repertorio más amplio.
El compromiso óseo debe hacer pensar
en sarcomas como primera posibilidad y los
síntomas reumatológicos en linfoma
o leucemia. El compromiso cutáneo y
de partes blandas puede tener su origen en
variados tumores de órganos internos,
o no ser manifestación de neoplasia.
El compromiso retroorbitario
del paciente recién expuesto, requiere
un examen oftalmológico que descarte,
por ejemplo, un rabdomiosarcoma derivado del
músculo estriado de la musculatura
oculomotora, que también debuta con
proptosis de rápida evolución
asociada a edema palpebral.
Los linfangiomas,
de crecimiento lento, pueden producir deformidad
estética y neuropatía óptica
compresiva, al igual que el glioma del nervio
ótico que debuta con un déficit
de agudeza visual pudiendo existir papiledema
o atrofia del nervio óptico. La leucemia
puede presentarse como proptosis aguda uni
o bilateral, sobre todo la variante mieloblástica
aguda.
Por último,
frente a equimosis palpebral, como el paciente
del caso clínico, en un niño
previamente sano, debe siempre descartarse
el trauma, fractura de base de cráneo
y el maltrato.
Tratamiento
El tratamiento
se basa en el protocolo creado por el PINDA
(Programa Nacional de Drogas Antineoplásicas
en el Niño), el que tiene como eje
un tratamiento integral del paciente en torno
a 3 esferas, Cirugía, Quimioterapia
y Radioterapia, dependiendo el abordaje, de
la etapificación del tumor según
el Sistema Internacional de Estadificación
del Neuroblastoma (Tabla 3), lo que permite
clasificar a los pacientes en diferentes grupos
de riesgo al adecuar el estadio con la edad
del paciente y las manifestaciones clínicas
del tumor (sintomático/ no sintomático).
Conclusiones
La clínica
y la sospecha médica son importantes
herramientas para el diagnóstico. Aún
así, deben acompañarse de técnicas
que lo confirmen, dado que hay hallazgos que
requieren un manejo precoz, tales como el
cáncer, cuyo pronóstico y sobrevida
dependen de la prontitud de la asistencia
médica.
Referencias
- Duarte J., Ruano
J., Calderón C. Neuroblastoma.
Oncología Médico Quirúrgica
Pediátrica Editorial McGraw
Hill interamericana editores S.A.
2001; 20: 186-201.
- López-Ibor
B, Moreno L. Tumores de la cresta
neural. En Hematología y Oncología
Pediátricas. Madero López
L. y Muñoz Villa A. (eds).
1997; 501-520.
- Parra R. y García
C. Caso
clínico-radiológico
para diagnóstico. Rev.
Chil. Ped 2000; 71(4): 347-349. ISSN
0370-4106. [Citado 10 de Julio 2006]
Disponible en internet: http://www.scielo.cl/scielo.php?script=sci_arttext&pid=S0370-41062000000400011&lng=es&nrm=iso&tlng=es
- Pose G. Estudio
de las imágenes en el diagnóstico
del cáncer infantil. Rev.
chil. ped. 2001; 72(2) 150-153. ISSN
0370-4106. [Citado 10 de Julio 2006]
Disponible en internet: http://www.scielo.cl/scielo.php?script=sci_arttext&pid=S0370-41062001000200012&lng=es&nrm=iso&tlng=es
- Protocolo Nacional
Neuroblastoma 1998. PINDA CHILE.
- Rostion C.G.,
Jáuregui L. Neuroblastoma:
Forma de presentación y probabilidad
de resección quirúrgica.
Rev. Ped. Elec. 2005, Vol 2, N°
2. ISSN 0718-0918 [Citado 10 de Julio
2006] Disponible en internet: http://www.revistapediatria.cl/vol2num2/11.htm
|